Type 1 galactosemia is a rare genetic disease that can be life-threatening in newborns and can lead to lifelong cognitive, neurological, and speech complications, as well as primary ovarian insufficiency in girls and women.[1] Type 1 galactosemia is caused by mutations in the GALT gene, leading to a profound deficiency of the GALT enzyme. This enzyme is required to break down galactose, a simple sugar endogenously produced by the body that is also found in dairy and other foods including breast milk.[1,2] A diet that restricts lactose and galactose is lifesaving in the first weeks of life. However, dietary intervention is inadequate in preventing long-term complications of the disease. The body produces significant, continuous levels of galactose, which is hypothesized to contribute to the long-term complications.[1]
Galactose plays a key role in the production of glycolipid and glycoproteins and is also involved in energy production, protein utilization and transport, and tissue synthesis. Without sufficient functional GALT enzyme, patients with galactosemia can experience excessive blood and tissue concentrations of galactose and its metabolites including galactose 1-phosphate (Gal-1p) and galactitol. This accumulation can be toxic and may harm organs and tissues throughout the body.[2,3]
This article provides an overview of the etiology, current standard of care, and the unmet need associated with Type 1 galactosemia.
Type 1 Galactosemia Etiology
Galactosemia was first described in 1908.[4] Patients with this disorder cannot convert the common carbohydrate galactose to glucose, which the body uses in many metabolic functions.[2,3] The underlying genetic cause of this disorder was not identified until 1956, when the galactose-1-phosphate uridylyltransferase (GALT) enzyme was implicated.[4] Galactosemia caused by mutations in the GALT gene is designated Type 1 galactosemia.
In total, four types of galactosemia are known, based on the enzyme deficiencies necessary for galactose metabolism; each is a result of mutations in a different gene.[2] In Type 1 galactosemia, mutations in the GALT gene hamper production of the GALT enzyme. In Type 2, a deficiency of galactokinase (GALK) is present, the result of mutations in the GALK1 gene; in Type 3, a deficiency of UDP-galactose 4-epimerase (GALE) is responsible (mutations in the GALE gene); and in Type 4, galactose mutarotase (GALM) production is reduced (mutations in the GALM gene).[2]
Focusing on Type 1 Galactosemia
It is important to remember that Type 1 galactosemia is not monolithic; there are three designated phenotypes.[1] The GALT gene may be affected by any one of more than 300 different variant mutations,[2] and individual patients with Type 1 galactosemia may therefore demonstrate different levels of GALT enzyme activity. In patients with classic galactosemia (the most severe form), GALT enzyme activity is virtually nonexistent (often below 1% that of unaffected individuals). The most common genetic mutations associated with classic galactosemia in patients of European ancestry are Q188R, K285N, and L195P.[1] These patients require lifelong adherence to the current standard of care (a galactose-restricted diet), and often still develop severe lifelong complications from the disease. This is believed to result from the body’s natural production of galactose (1–2 g/day).[1]
In the phenotype known as “clinical variant” galactosemia, patients’ GALT enzyme activity ranges from 1% to 15% of that seen in unaffected individuals.[1] The most common genetic mutation associated with clinical variant galactosemia is S135L.[1] Patients with clinical variant galactosemia remain at risk for severe early symptoms.[1] They may also be at risk for developing lifelong complications of the disease. Based on the international clinical guidelines, a galactose-restricted diet is recommended for patients with clinical variant galactosemia who present with less than 10% of GALT enzyme activity and/or have the S135L mutation.[5] Insufficient information is known about patients with between 10% and 15% GALT enzyme activity; therefore, the clinical guidelines do not advise whether these patients should be treated.[5]
Finally, patients with Duarte galactosemia (also referred to as “biochemical variant” galactosemia) demonstrate an average GALT enzyme activity of 25% of that seen in unaffected individuals,[1] which allows most patients to remain asymptomatic. These patients have one allele with a severe GALT pathogenic mutation and the other allele with alterations that include N314D polymorphism.[1] These patients generally have normal development and no long-term complications.[6,7] Some families of children with Duarte galactosemia choose to restrict dairy in the first year of life[6] but the current international guidelines do not recommend this.
Impaired Metabolism and Its Effects
After ingestion of lactose from milk and dairy products, this carbohydrate is broken down in the intestines into galactose and glucose.[8] Galactose is necessary for the galactosylation of proteins, ceramides involved in myelin sheath synthesis, and other substances.[3] Patients with classic galactosemia can convert lactose into galactose, but they cannot complete the metabolic conversion of galactose to glucose as a result of their lack of GALT enzyme activity.[2] The metabolism of galactosemia is described by the Leloir pathway [Figure 1].[2]
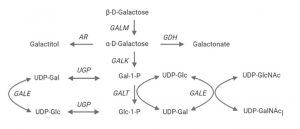
Figure 1. The Leloir pathway of galactose degradation. Abbreviations: AR = aldose reductase; Gal-1-P = galactose-1-phosphate; GALE = UDP-galactose 4-epimerase; GALK = galactokinase; GALM = galactose mutarotase; GALT = galactose 1-phosphate uridylyltransferase; GDH = galactose dehydrogenase; Glc-1-P = glucose-1-phosphate; UDP-Gal = uridine diphosphate-galactose; UDP GalNAc = UDP-N-acetylgalactosamine; UDP-Glc = uridine diphosphate-glucose; UDPGlcNAc = UDP-N-acetylglucosamine; UGP = UDP-glucose pyrophosphorylase. Modified from Delnoy et al 2021.[2]
The excess galactose accumulates in cells, along with two of its metabolites (Gal-1p and galactitol) due to the GALT enzyme deficiency.[1] The oversupply of these molecules can be toxic and may contribute to a rapid cascade of serious lifelong developmental, motor and coordination, physiologic, and neurologic complications.[2] The basis for the downstream effects of galactosemia are not fully understood, but oxidative stress on organ tissues is theorized to play a role.[2]
It is of the utmost importance to immediately decrease the accumulation of galactose and its metabolites in cells and tissues via dietary interventions.[5] Such action can lower the risk of life-threatening complications and death in newborns.[1]
For perspective, one glass of milk may contain 4.8 g of galactose, and although a galactose-restricted diet may reduce the amount of consumed galactose to 0.054 g/day or less,[9] dietary restriction of galactose alone does not fully protect patients from the complications of Type 1 galactosemia.[9] According to Berry,[1] the body naturally produces about 1–2 g of galactose per day, regardless of diet. Thus, endogenous production of galactose may be the main contributor to the complications of galactosemia once diet is controlled.[1] Type 1 galactosemia and lactose intolerance are very different. They may both require dietary modification, but this alone will not prevent the lifelong effects of galactosemia.
Prevalence
Type 1 galactosemia, an autosomal recessive genetic disorder, is rare, and prevalence estimates may be based on limited studies with gaps in the data. The prevalence of classic galactosemia is reported to be approximately 1:30,000–60,000 newborns in the U.S.[1]. Confirmed estimates of the prevalence of the clinical variant subtype are lacking; however, one estimate of the prevalence of individuals from South Africa with the S135L/S135L mutation genotype is 1:20,000.[1] The prevalence of Duarte galactosemia is likely much more common, with a prevalence of 1:4,000 infants of European ancestry, according to Pyhtila and colleagues.[10] .
Diagnosis
Although infants with Type 1 galactosemia may seem healthy at birth, clinical signs of this disorder are often seen as soon as the infant is exposed to mother’s milk or milk-based formula. Newborn screening is the bedrock for identifying patients with a GALT gene mutation in the U.S. and many other countries.[10]
Galactosemia was first added to the newborn screening panel in the U.S. in 1963, and it has been required by all states since 2004.[10] Newborn testing is designed to screen for 100% of babies affected by classic and clinical variant galactosemia.[1] Screening is not immediately performed upon delivery (the methodology of testing and timing of newborn blood collection for screening varies among states);[10] therefore, infants are almost always exposed to breast milk or galactose-containing formula prior to diagnosis.
Because Type 1 galactosemia symptoms in newborns may first appear as common complications, including jaundice and poor feeding, it is difficult to identify immediately without newborn screening.[10] Newborn screening allows for early detection so infants can be switched to safe formula as soon as possible. This is imperative, as the mortality rate for infants who do not switch to a non-milk-based formula is about 75%.[3]
A positive screening result requires confirmatory biochemical and/or genetic testing to confirm low erythrocyte GALT enzyme activity or high levels of galactose and/or Gal-1p. False-positive results on newborn screening are common; one cause may be due to detection of infants who carry one mutated allele.[10] Another potential reason for false-positive test results may be degradation of enzymes prior to analysis.[11] Subsequent molecular genetic screening can identify the mutation and verify a diagnosis.[1]
Presentation
Once a newborn ingests breast milk or conventional formula, both rich sources of galactose, symptoms appear rapidly. These typically include signs of liver damage (elevated liver enzymes, jaundice, and hepatomegaly), problems in feeding, failure to thrive (with vomiting), sepsis (most commonly caused by Escherichia coli), and cataracts (Table 1). Less commonly seen are lethargy, hypotonia, and renal tubular impairment.[12] If a galactose-restricted diet is started within 1 week of birth, the risk of fatal or severe/significant neonatal complications drops rapidly (odds ratio, 32%).[1,12]
TABLE 1: ACUTE NEONATAL ILLNESS IN CLASSIC AND CLINICAL VARIANT GALACTOSEMIA*: INTERNATIONAL PATIENT REGISTRY RESULTS[12]
| Symptom/Sign | Frequency (%) |
| Liver damage (elevated liver enzymes) | 70% |
| Bleed diathesis | 42% |
| Altered mental state | 29% |
| Infection | 27% |
| Cataracts | 26% |
| Hypoglycemia | 25% |
*Eighty-three percent of the study population had GALT enzyme activity of ≤ 1%; 17% had GALT enzyme activity > 1% to ≤ 10% of normal.
Galactosemia Family Story: Allison and Brooks Woodfin
Lifelong Complications
Even with early dietary modification, patients with classic and clinical variant galactosemia are at risk for cognitive, neurological, and speech complications, as well as well as primary ovarian insufficiency in females (Table 2). These complications are highly variable from one patient to another, although several issues may be inter-related (e.g., developmental delay with speech or motor problems). Figure 2 illustrates the natural history of Type 1 galactosemia across the patients’ lifespans.
Figure 2. Type 1 galactosemia natural history. Note: the clinical phenotype can vary significantly. *83% of females experiencing premature ovarian insufficiency (i.e., 80% of females with type 1 galactosemia). Adapted from Rubio-Gozalbo et al.[12]
Natural History Figure 2 Galactosemia

A registry of patients with Type 1 galactosemia (83% with the classic subtype, and 17% with the clinical variant subtype) found that speech and language disorders are apparent in up to two-thirds of these patients.[12] Approximately half of the patients in the registry showed developmental delays (including motor and cognitive complications).[12]
Neurologic complications, including dystonia, tremor, and seizures, are also reported in more than 50% of patients. These symptoms tend to present in the late teens and in young adults.[12]
Psychiatric and behavioral problems, which occur in 44% of patients, include anxiety, depression, autism spectrum disorder, and attention deficit hyperactivity disorder.[12]
Gonadal disorders are also common in patients in the registry. The incidence of primary ovarian insufficiency was reported in 80% of females, of which more than 80% required hormone replacement therapy.[12] For those females who seek to become pregnant and have trouble conceiving, fertility treatment options may be necessary (including oocyte donation).[5]
Delayed growth was also reported, though final attained heights were within average parameters.[1]
Low bone-mineral density (BMD) was recorded in 26% of patients in the galactosemia registry, two-thirds of whom were female.[12]
TABLE 2: LONG-TERM COMPLICATIONS IN CLASSIC AND CLINICAL VARIANT GALACTOSEMIA*: PATIENT REGISTRY RESULTS[12]
| Symptom/Sign | Frequency (%) |
| Primary ovarian insufficiency (age, < 40 yr) | 80% |
| Language and speech disorders | 66% |
| Developmental delay in infancy/childhood Language delay Motor and cognitive Cognitive only |
52% 78% 50% 39% |
|
Neurologic complications Tremor Ataxia Seizures |
52% 31% 12% 8% |
| Psychiatric/behavioral problems | 44% |
| Spontaneous puberty in females | 51% |
| Induced puberty† | 48% |
| Osteopenia (reduced bone mineral density and higher risk of bone fracture) | 26% |
| Non-neonatal development of cataracts | 9% |
*Study included 509 patients from 15 countries, aged 0 to 65. Eighty-three percent of the study population had GALT enzyme activity of ≤ 1%; 17% had GALT enzyme activity > 1% to ≤ 10% of normal.
†Rate given for females; delayed puberty occurred in 5% of males.
Galactosemia Family Story: Megan and Ava Lilja
Galactosemia Family Story: David and DJ Trainor
Standard of Care and Management
Upon diagnosis of classic galactosemia (or GALT enzyme activity ≤10% of normal range; Gal-1p concentration ≥10 mg/dL), initiation of immediate dietary modification is the standard of care.[5] Specialized soy formulas or galactose-free formula may be used to feed these infants. It is also important to note that some medications may contain lactose, and these may need to be avoided. For a newborn, dietary modification can mean avoidance or reversal of liver failure and possible death.[13]
Upon initial diagnosis, consultation with specialists with experience in inherited metabolic disorders is highly recommended.[1] Furthermore, evaluation of the infant for developmental issues, neurologic function, feeding and nutritional status, cataracts, and infection prevention suggests that a team-based approach to care is optimal.[1]
Ongoing management of patients with galactosemia focuses on managing lifelong complications. Because of the body’s endogenous production of galactose,[1] accumulation of galactose and its toxic metabolites may continue to pose a risk for long-term tissue damage. Clinical guidelines recommend annual testing of red blood cell levels of Gal-1p, until individual baseline levels have been established[5]; this can also serve to monitor any galactose intake.
For many patients, ongoing management includes occupational, physical, speech, and feeding therapy. In patients with cognitive or language delays, or in patients exhibiting signs of motor dysfunction, individual therapy can be customized for the best patient care.[1,5]
To counter reduced BMD in patients with galactosemia, calcium and vitamin D supplementation is often recommended.[1]
There are currently no approved therapies to correct deficiencies in GALT enzyme activity. However, development of potential treatments, including gene therapy, mRNA therapy, and other approaches that may work to prevent the toxic accumulation of galactose and its multiple metabolites in the blood, are currently in development.[2]
Researchers believe that treatments to address the underlying deficiency in GALT enzyme activity, delivered soon after diagnosis, may someday give patients the best chance to avoid the lifelong clinical complications of Type 1 galactosemia.[2]
Galactosemia Resources
The Galactosemia Foundation The organization that advocates for people with galactosemia and their families. The Galactosemia Foundation helps provide families information about galactosemia and facilitates networking among families, clinicians, and researchers.
Galactosemia Network (GalNet). The international network for professionals focused on the advancement of research, diagnosis, treatment, and follow-up care of patients with galactosemia.
National Organization for Rare Diseases (NORD). A patient advocacy organization dedicated to individuals with rare diseases and the organizations that serve them.
GENE-THERAPY RESOURCES
American College of Molecular Genetics. A professional society representing the interests of the entire medical genetics team, including clinical geneticists, clinical laboratory geneticists, and genetic counselors.
American Society of Gene & Cell Therapy. This organization (comprised of scientists, physicians, and patient advocates) seeks to advance knowledge, awareness, and education leading to the discovery and clinical application of genetic and cellular therapies to alleviate human disease.
American Society of Human Genetics. A professional membership organization for specialists in human genetics, with approximately 8,000 members.
Society for Inherited Metabolic Disorders. The Society aims to increase knowledge of and promote research in inborn errors of metabolism in humans and to stimulate interactions between clinicians and investigators in inborn errors of metabolism.
References
- Berry GT. Classic Galactosemia and Clinical Variant Galactosemia. 2000 Feb 4 [Updated 2021 Mar 11]. In Adam MP, Ardinger HH, Pagon RA, et al., editors GeneReviews®. Seattle (WA): University of Washington. Seattle: 1993-2021. https://www.ncbi.nlm.nih.gov/books/NBK1518/. Accessed July 30, 2021.
- Delnoy B, Coelho AI, Rubio-Gozalbo ME. Current and future treatments for classic galactosemia. J Personalized Med. 2021;11(2):75. https://doi.org/10.3390/jpm11020075
- Kotb MA, Mansour L, Shamma RA. Screening for galactosemia: Is there a place for it? Int J Gen Med. 2019:12:193-205. https://doi.org/10.2147/IJGM.S180706
- History of galactosemia. Galactosemia Foundation http://www.galactosemia.org/history-of-galactosemia. Accessed January 7, 2022.
- Welling L, Bernstein LE, Berry GT, et al. International clinical guideline for the management of classical galactosemia: diagnosis, treatment, and follow-up. J Inherit Metab Dis. 2017;40:171-176. https://doi.org/10.1007/s10545-016-9990-5
- Fridovich-Keil JL, Gambello MJ, Singh RH, et al. Duarte variant galactosemia. 2014 Dec 4 [Updated 2020 Jun 25]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020: https://www.ncbi.nlm.nih.gov/books/.
- Carlock G, Fischer ST, Lynch ME, et al. Developmental outcomes in Duarte galactosemia. Pediatrics. 2019;143(1):e20182516. https://doi.org/10.1542/peds.2018-2516
- Coelho AI, Rubio-Gozalbo ME, Vicente JB, et al. Sweet and sour: An update on classic galactosemia. J Inherit Metab Dis. 2017;40:325-342. https://doi.org/10.1007/s10545-017-0029-3
- Bosch AM. Classic galactosemia: Dietary dilemmas. J Inherit Metab Dis. 2011;34:257-260. https://doi.org/10.1007/s10545-010-9157-8
- Pyhtila BM, Shaw KA, Neumann SE, et al. Newborn screening for galactosemia in the United States: looking back, looking around, and looking ahead. JIMD Rep. 2015;15:79-93. https://doi.org/10.1007/8904_2014_302
- Pasquali M, Yu C, Coffee B, et al. Laboratory diagnosis of galactosemia: a technical standard and guideline of the American College of Medical Genetics and Genomics (ACMG). Gen Med. 2018;20:3-11. https://doi.org/10.1038/gim.2017.172
- Rubio-Gozalbo ME, Haskovic M, Bosch AM, et al. The natural history of classic galactosemia: Lessons from the Galnet registry. Orphanet J Rare Dis. 2019;14(86):1-11. https://doi.org/10.1186/s13023-019-1047-z
- Galactosemia. National Organization for Rare Diseases. Updated 2019. https://rarediseases.org/rare-diseases/galactosemia/. Accessed January 12, 2022.
