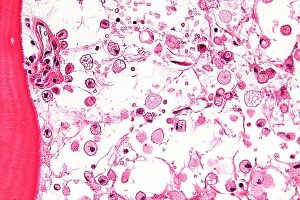
Micrograph of Gaucher disease, with cells that have the characteristic crumpled tissue paper-like cytoplasm.
Lysosomal storage disorders (LSDs) are a group of about 50 genetic diseases caused by defects in lysosomal proteins or lysosomal related-proteins, which results in dysfunction of lysosomes. Most LSDs are caused by the genetic absence of a single catabolic enzyme, causing accumulation of the enzyme’s substrate within the lysosome. Over time, tissue-specific substrate accumulations result in a spectrum of symptoms and disabilities that vary by LSD.
Almost all LSDs (with Hunter syndrome [MPS II] and Fabry disease being two exceptions) are autosomal recessive disorders. This means both parents must carry the abnormal gene that prevents the body from making an enzyme with normal activity. Although the different types of LSDs are rare individually, taken together LSDs as a whole affect roughly 1 in 7,700 births, making them a more common health problem.
Symptoms and Affected Areas
All LSDs are progressive, which makes diagnosing and starting treatment of the highest importance. The rate of progression, severity of symptoms and organs affected vary among the various LSDs and even within each individual disorder type. LSDs can affect different body organs or systems that include:
• Bones and joints
• Central nervous system
• Eyes
• Heart
• Kidneys
• Lungs
• Spleen
• Liver
• Skin
Although the signs and symptoms vary from disease to disease, symptoms of LSDs occur because of an enzyme deficiency that inhibits the ability of the lysosomes present in each of the body’s cells to perform their normal function. The lysosomes function as the primary digestive units within cells. Their function is to break down complex components into simpler ones. Each cell has hundreds of lysosomes that degrade complex cellular components such as proteins (substrates) into simpler components. When this process does not take place, the substrate begins to accumulate in the cells.
Like most rare diseases, LSDs pose patients with several challenges, including a proper and timely diagnosis. It is often difficult for physicians to diagnose LSDs because symptoms vary among the different types and individual LSDs are rare. Physicians can usually confirm a diagnosis when they recognize a pattern of symptoms, but this process may take years while clinicians rule out other conditions. Specialized laboratories offer specific diagnostic tests that can confirm or rule out an LSD, but even the number of tests available can sometimes confuse the diagnosis path.
Management and Treatments
In the past 10 years, there have been significant developments in the treatment of lysosomal storage disorders as a number of therapies have been approved by the FDA and the number of clinical trials has grown considerably. This has been motivated partly by the incentives provided by orphan drug regulations that provide marketing exclusivity and other commercial benefits, but also because of medical breakthroughs and increased understanding of LSDs.
Although the new approved therapies are not treatments for the various disorders, they can stabilize organ functions or slow the progression of the disease. In addition, because of the broad clinical variability of these conditions, the benefit of a therapy cannot be predicted in an individual patient. The Table below highlights some of the new therapies have been approved for lysosomal storage disorders and some that have received an orphan drug status.
Some of the most common lysosomal storage diseases and a few of their characteristic signs and symptoms are as follows:
Batten Disease:
Batten disease is the juvenile form of a group of progressive neurological disorders known as neuronal ceroid lipofuscinoses (NCL). It is characterized by the accumulation of a fatty substance (lipopigment) in the brain, as well as in tissue that does not contain nerve cells. Batten disease is marked by rapidly progressive vision failure (optic atrophy) and neurological disturbances, which may begin before eight years of age. Occurring mostly in families of Northern European Scandinavian ancestry, the disorder affects the brain and may cause deterioration of both intellect and neurological functions.
Cystinosis:
The early signs of this disorder typically involve the kidneys and the eyes. Excessive storage of the amino acid cystine in all cells of the body results in impaired kidney function, increased sensitivity to light, and marked growth retardation. There are infantile (the most common and most severe), juvenile, and adult forms, each with associated symptoms.
Fabry Disease:
The symptoms of Fabry disease usually begin during early childhood or adolescence. However, they may not become apparent until the second or third decade of life. Early symptoms include episodes of severe burning pain in the hands and feet. Other early signs may include a decrease in sweat production, discomfort in warm temperatures, and the appearance of a reddish to dark blue skin rash, especially in the area between the hips and knees. These skin lesions may be flat or raised, and some people may not have them at all.
Gaucher Disease Types I, II, and III:
Gaucher disease is the most common type of lysosomal storage disorder. Researchers have identified three distinct types of Gaucher disease based upon the absence (type I) or presence and extent of (types II and III) neurological complications. Most affected individuals have type I. These patients may experience easy bruising, chronic fatigue, and an abnormally enlarged liver and/or spleen (hepatosplenomegaly). Gaucher disease type II occurs in newborns and infants, and is characterized by neurological complications that may include involuntary muscle spasms, difficulty swallowing and the loss of previously acquired motor skills. Gaucher disease type III appears during the first decade of life. Neurological complications may include mental deterioration, an inability to coordinate voluntary movements, and muscle spasms of the arms, legs, or entire body.
Glycogen Storage Disease II (Pompe Disease):
Pompe disease has an infantile form and a delayed onset form. The delayed onset form may be further broken down into a childhood form and a juvenile/adult form. Patients with the infantile form are the most severely affected. Although these infants usually appear normal at birth, the disease presents within the first two to three months with rapidly progressive muscle weakness, diminished muscle tone (hypotonia) and a type of heart disease known as hypertrophic cardiomyopathy. Feeding problems and respiratory difficulties are common. The childhood form presents during infancy or early childhood. Motor milestones may be delayed and some symptoms may resemble muscular dystrophy. The cardiac enlargement that is often present in the infantile form is seldom seen in the childhood form.
The juvenile/adult form presents between the first and seventh decades as a slowly progressive muscle weakness or with symptoms of respiratory insufficiency. There is no cardiac involvement with this form.
GM2-Gangliosidosis Type I (Tay Sachs Disease):
Two main forms of Tay Sachs disease exist: the classic or infantile form and the late-onset form. In individuals with infantile Tay Sachs disease, symptoms typically first appear between three and five months of age. These may include feeding problems, general weakness (lethargy), and an exaggerated startle reflex in response to sudden loud noises. Motor delays and mental deterioration are progressive. In individuals with the late-onset form, symptoms may become apparent anytime from adolescence through the mid-30s. The infantile form often progresses rapidly, resulting in significant mental and physical deterioration. A characteristic symptom of Tay Sachs disease, which occurs in 90 percent of cases, is the development of cherry red spots in the backs of the eyes. Symptoms of late-onset Tay Sachs disease vary widely from case to case. This disorder progresses much more slowly than the infantile form.
GM2-Gangliosidosis Type II (Sandhoff Disease):
The first symptoms of Sandhoff disease typically begin between the ages of three and six months. The disease is clinically indistinguishable from GM2-Gangliosidosis Type I.
Metachromatic Leukodystrophy:
Early signs and symptoms may be vague and gradual, making this disorder difficult to diagnose. Unsteadiness when walking is often the first symptom observed. Occasionally, the earliest symptom is developmental delay or deteriorating school performance. Over time, symptoms may include marked spasticity, seizures, and profound mental retardation.
Mucolipidosis Types I, II/III and IV:
Mucolipidosis I, also known as sialidosis, has juvenile and infantile forms (sialidosis type I and sialidosis type II). Sialidosis type I usually becomes apparent during the second decade of life with the advent of sudden involuntary muscle contactions, the appearance of red sopts (cherry-red macules) in the eyes, and/or other neurological findings. Sialidosis type II may begin during infancy or later and is characterized by the same visual characteristics as sialidosis type I, as well as other symptoms such as mildly coarse facial features, skeletal malformations, and/or mild mental retardation. Symptoms of ML II, also known as I-cell disease, typically become apparent during infancy and include abnormalities of the skull and face, growth failure, and/or mental retardation.
Type III, also known as pseudo-Hurler disease, is characterized by stiffness of the hands and shoulders with later development of carpal tunnel syndrome, deterioration of hip joints, scoliosis, and short stature. ML IV is characterized by mental retardation, greatly reduced ability in the acquisition of skills requiring the coordination of muscular and mental activities, corneal clouding, retinal degeneration, and diminished muscle tone.
Mucopolysaccharide Storage Diseases (Hurler Disease and variants, Hunter, Sanfilippo Types A,B,C,D, Morquio Types A and B, Maroteaux-Lamy and Sly diseases):
The MPS diseases are caused by disturbances in the normal breakdown of complex carbohydrates known as mucopolysaccharides. All of the MPS diseases have certain characteristics in common. These include deformities of the bones and joints that interfere with mobility and often cause osteoarthritis, especially of the large, weight-bearing joints. All of the MPS diseases except Sanfilippo disease interfere with growth, causing short stature.
Niemann-Pick Disease Types A/B, C1 and C2:
Niemann-Pick disease is a group of inherited disorders related to fat metabolism. Certain characteristics common to all types include enlargement of the liver and spleen. Children with Niemann-Pick disease also experience progressive loss of motor skills, feeding difficulties, progressive learning disabilities, and seizures.
Resources
Acid Maltase Deficiency Association (Pompe)
National Fabry Disease Foundation
To learn more about rare lysosomal storage disorders, visit https://checkrare.com/diseases/lysosomal-storage-disorders/
