Niemann-Pick disease type C (NPC) is a disabling, lysosomal storage disorder that has been diagnosed prenatally, neonatally, during childhood, and even into adulthood.[1,2] This very rare genetic disorder is marked by progressive motor dysfunction and a highly variable symptom profile and onset of symptoms.[3] It can result in the patient’s death soon after birth[4] or manifest as a chronic disorder with symptoms worsening slowly over time.[2]
The underlying, principal abnormality is the cell’s inability to adequately move fatty molecules (e.g., cholesterol and lipids) out of the cell’s lysosomes, resulting in accumulations in the lysosomes and late endosomes.[1] This leads to fatty substances building up in many organ systems throughout the body.[1,5] This dysfunction has been associated with mutations in one of two genes (NPC1 or NPC2).[1]
In patients who are diagnosed beyond the perinatal period (>2 months), progressive psychiatric dysfunction and neurologic abnormalities may be observed.[1] In younger patients (<15 years old), the earlier symptoms tend to be associated with visceral organ involvement.[1,6]
Diagnosis may be delayed in patients with NPC,[7] as numerous other disorders need to be considered. Genetic testing will generally yield the diagnosis.[7]
Management of NPC largely consists of treating patients’ individual symptoms such as urologic deterioration, seizures, hypersalivation and drooling, spasticity, bowel dysfunction, and behavioral or psychiatric issues.[1] Although miglustat was approved for the treatment of NPC by the European Medicines Agency in 2006,[8] it has not been approved for this indication by the U.S. Food and Drug Administration.[9] The drug inhibits the glycosphingolipid (GSL) synthesis, which seems to slow accumulation of this lipid in brain tissue.[9] As a result, it may slow the development of neurologic symptoms associated with NPC.[10]
Multidisciplinary care management may be the best approach to helping patients with NPC (and their families or caregivers), because NPC affects different organs, muscle tone, and mental status.[1]
The prognosis for the disease can be poor for infants (<2 months) demonstrating symptoms just after birth (like cholestasis, hepatomegaly, or splenomegaly), but other patients may have mild disease that is not diagnosed until they are adults.[1] For those expressing long-term, severe symptoms, death may result from neurologic decline and other life-threatening complications of NPC.[11]
Elizabeth Berry-Kravis, MD, PhD: Overview of Niemann-Pick Type C
EPIDEMIOLOGY AND PATHOPHYSIOLOGY
The incidence of NPC is very low—affecting approximately 1 baby in every 100,000 live births.[11] Patients with NPC are categorized by age of onset: (1) neonatal; (2) early infantile onset (<2 years); (3) late infantile onset (2-6 years); (4) juvenile onset (6-15 years); or (5) adolescent/adult onset (>15 years).[1,11] One international guideline further associates these ages of onset with primary manifestations: early infantile (visceral-neurodegenerative form), late-infantile/juvenile onset (neurodegenerative form), and adult-onset (psychiatric-neurodegenerative form).[1]
According to a U.K. population analysis, patients with late-infantile onset and juvenile onset seem most common, with an average age at the time of diagnosis of 4.6 and 11.5 years, respectively.[11] One 2006 case report described an NPC diagnosis in a 68-year-old woman.[2] Consensus clinical management guidelines for NPC confirms that although late-infantile and juvenile onset disease combine for the majority of cases, adolescent/adult-onset accounts for upwards of 30% of the patient population.
Disease prevalence does not seem to be higher in any specific region of the world[1]; however, some epidemiologic evidence suggests that the Hispanic populations of southern New Mexico and Colorado seem to be at higher incidence.[12] The only risk factor that seems confirmed by evidence is a familial history (specifically, in a parent or sibling) of NPC.[3]
Research has isolated the underlying cause of NPC to be autosomal recessive mutations in either the NPC1 or NPC2 genes, the former being responsible for approximately 95% of cases.[1] NPC1 encodes for the NPC1 protein, which is found in the lysosomal membrane. It is responsible for cholesterol and sphingolipid transport and metabolism within the cell. The primary mutation to NPC1 destabilizes the protein, preventing its function, causing the accumulation of these fatty substances.[13] In rare cases, the NPC2 gene is involved; NPC2 encodes for a small protein in the lysosome that attaches to cholesterol and transports it to the NPC1 protein for metabolism. Dysfunction in this gene also results in accumulation of lipids in the lysosome and endosome.[13]
SIGNS AND SYMPTOMS
As indicated, signs and symptoms may vary by age of onset, and they may also vary within each age group (Table 1).[1,3] The symptoms of NPC may be highly variable from patient to patient.
Findings in the youngest NPC patients (prenatal/perinatal; <2 months) are primarily liver disease with prolonged cholestatic jaundice, hepatomegaly and splenomegaly, and in some cases acute liver failure, with or without pulmonary disease. In 8-9% of cases, liver failure may develop. Lack of muscle tone (hypotonia) and failure to thrive may be evident.[1]
TABLE 1: SUMMARY OF CLINICAL SIGNS AND SYMPTOMS IN NIEMANN-PICK SYNDROME, BY AGE OF ONSET[1]
| Age at Onset | Systemic Manifestations | Neurological/Psychiatric Manifestations |
| Pre-/perinatal (<2 mo) |
Fetal ascites/hydrops Hepatosplenomegaly Cholestatic jaundice Thrombocytopenia Pulmonary disease Liver failure Failure to thrive |
Hypotonia |
| Early infantile (2–24 mo) |
Hepatosplenomegaly or splenomegaly (isolated or with neurologic manifestations)
Prolonged neonatal jaundice
|
Central hypotonia Delayed developmental motor milestones, speech delay
Dysphagia, spasticity VSGP |
| Late infantile (2–6 yr) |
Hepatosplenomegaly or splenomegaly (isolated or with neurologic manifestations)
History of prolonged neonatal cholestatic jaundice |
Developmental delay/regression, speech delay Clumsiness, frequent falls Progressive ataxia, dystonia, dysarthria, dysphagia Seizures (partial/generalized) Cataplexy VSGP Hearing loss |
| Juvenile (6–15 yr) | Hepatosplenomegaly or splenomegaly (isolated or with neurologic manifestations) |
Poor school performance, learning disability Loss of language skill Progressive ataxia, dystonia, dysmetria, dysarthria, dysphagia VSGP Gelastic cataplexy Seizures Behavioral problems |
|
Adolescent/adult (>15 yr) |
Splenomegaly (often not present; isolated in very rare cases) |
Cognitive decline, dementia, learning disability Psychiatric signs: Schizophrenia (psychosis), depression Clumsiness, progressive motor symptoms, tremor, ataxia, dystonia/dyskinesia, dysarthria, dysphagia VSGP |
|
VSGP = vertical supranuclear gaze palsy. Adapted from Geberhiwot et al 2018[1] with permission. |
||
FIGURE 1: CLASSIFICATION AND NEUROLOGIC INVOLVEMENT OF NIEMANN-PICK TYPE C DISEASE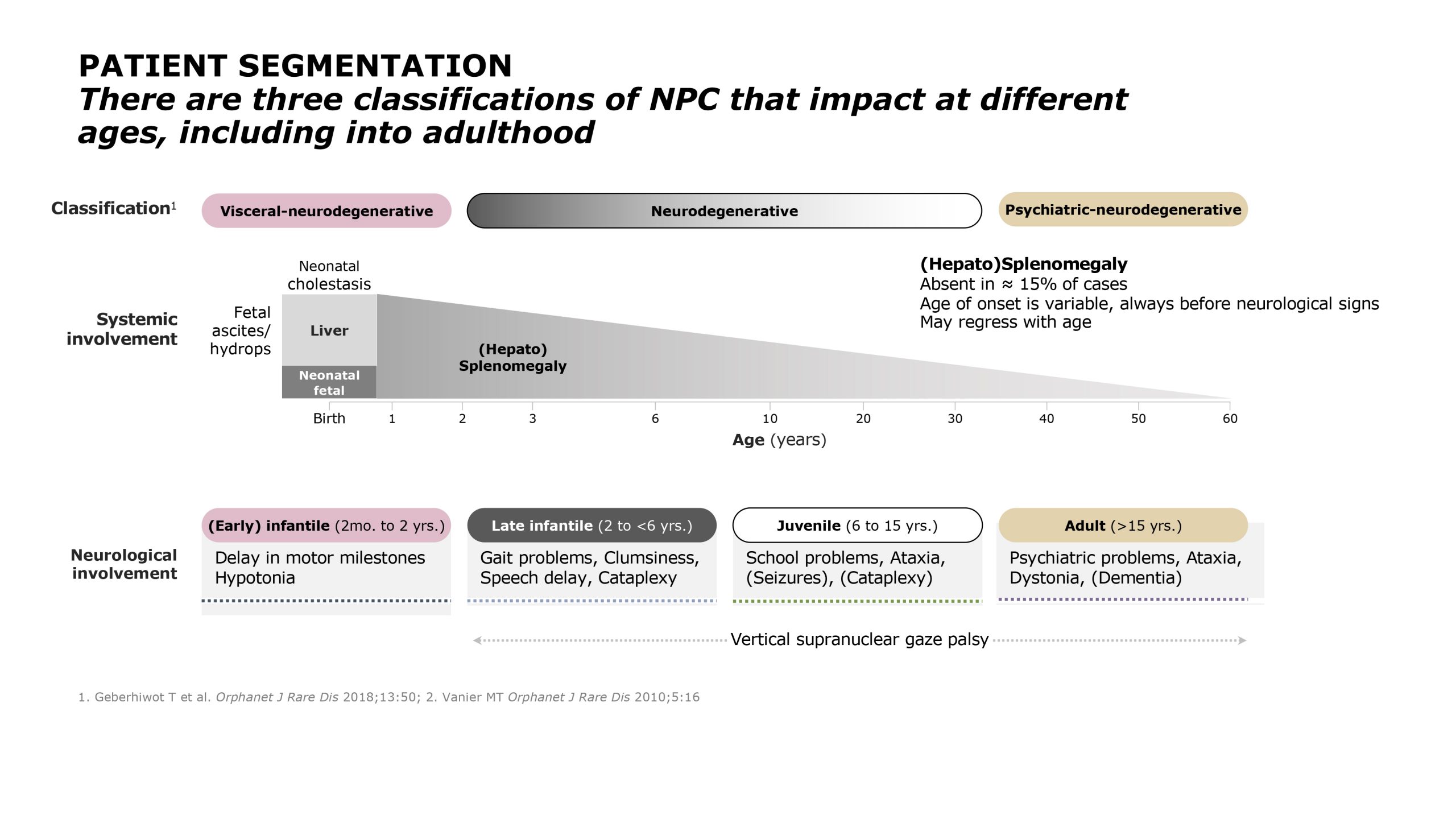
In the early-infantile NPC group (2 months to <2 years), enlarged liver and/or spleen are present, along with jaundice. Neurologically, hypotonia and failure to meet developmental milestones are often observed, including in the areas of motor development and coordination; however, the ability to communicate is not universally affected at this point.[1] One of the most specific signs of NPC is easy to overlook: most patients demonstrate vertical supranuclear gaze palsy (VSGP), or the impairment of the ability to look upward or downward rapidly.[1,7] Affected children make sudden abnormal head movements to compensate for this loss. Another specific sign is gelastic cataplexy.[3] Cataplexy is characterized by a sudden loss of muscle tone and strength that can cause sudden head drop, a weak, rubbery sensation in the legs, or in severe cases, collapse. Cataplexy is often caused by strong emotions, typically laughter, in individuals with NPC (gelastic cataplexy).[14]
Patients in the late-infantile NPC age group (2-6 years) may show the following signs: VSGP, gelastic cataplexy, speech delay, and epilepsy, and sensory hearing loss. Patients may have had a history of cholestasis neonatally. Some patients will exhibit enlargement of the visceral organs.[1]
In general, the first signs of NPC observed in children with juvenile-onset disease (6-15 years) are poor school performance; learning problems; and difficulties with gait, coordination, and motor skills (e.g., frequently falling). As a result of their motor coordination deterioration, patients may be characterized as “clumsy.” Speech abnormalities may also develop, which is associated with progressive ataxia. As with other patient cohorts, VSGP will likely be evident.[1] Children with NPC may also develop dysphagia, which could lead to aspiration pneumonia and general oral feeding issues.
Patients with adolescent/adult-onset NPC (>15 years) generally demonstrate signs of cognitive impairment, despite meeting all developmental milestones for younger children. Issues with complex thinking and reasoning could become apparent in older patients. Psychiatric symptoms (e.g., psychosis, depression) are often seen in this population. Some patients will present with splenomegaly, but others will not have any visceral signs. Patients in this age group will not display gelastic cataplexy or epileptic seizures.[1]
DIAGNOSIS
The diagnosis of NPC is challenging for a number of reasons: (1) pediatricians and family physicians will not likely have experience with NPC because it is a rare disease, (2) a high index of suspicion may be necessary to avoid misdiagnosis, (3) the frequency of psychiatric disorders may muddle the diagnosis, and (4) genetic testing is generally required to confirm the diagnosis.[1]
The delay in diagnosis from the onset of neurologic symptoms has been reported to be an average of 4 years.[7] The issues involved may be illustrated with adolescent/adult-onset NPC, owing to the variability in presentation and a differential diagnosis that could include schizophrenia, major depression disorder, and forms of dementia.[15] On the other hand, VSGP is a very common sign of NPC; it was reported in more than 70% of patients older than the late-infantile cohort (and dysarthria in >60%).[15]
Diagnosis is usually confirmed through the use of a combination of modalities, but the clinician would need a high index of suspicion to efficiently arrive at the diagnosis. The use of a specific screening tool may steer a physician toward considering NPC, but this suspicion index seems most effective in patients younger than 4 years.[3] As specified in diagnostic guidelines from Patterson and colleagues[7] (Figure 2), initial screening includes genetic testing, with subsequent sequencing of NPC1 and NPC2, in addition to biomarker testing. The following biomarkers have been proven useful in diagnosing NPC[7]:
- Cholesterol-oxidation products or oxysterols; cholestane-3β,5α,6β and 7-ketocholesterol are examples
- Lysosphingomyelin-509 and other lysosphingolipids
- Bile acids (e.g., 3β,5α,6β-trihydroxy-cholanoyl-glycine)
If the biomarker tests are positive, molecular gene sequencing studies are necessarty to confirm the diagnosis.[1,7] It should be noted that the filipin staining test was once considered the gold standard for diagnosing NPC. This test is not easily performed or accessible, and is now considered optional.[1]
FIGURE 2 REVISED NIEMANN-PICK DISEASE TYPE C DIAGNOSTIC ALGORITHM FOR THE USE OF BIOMARKERS AND GENETIC TESTING.[7]
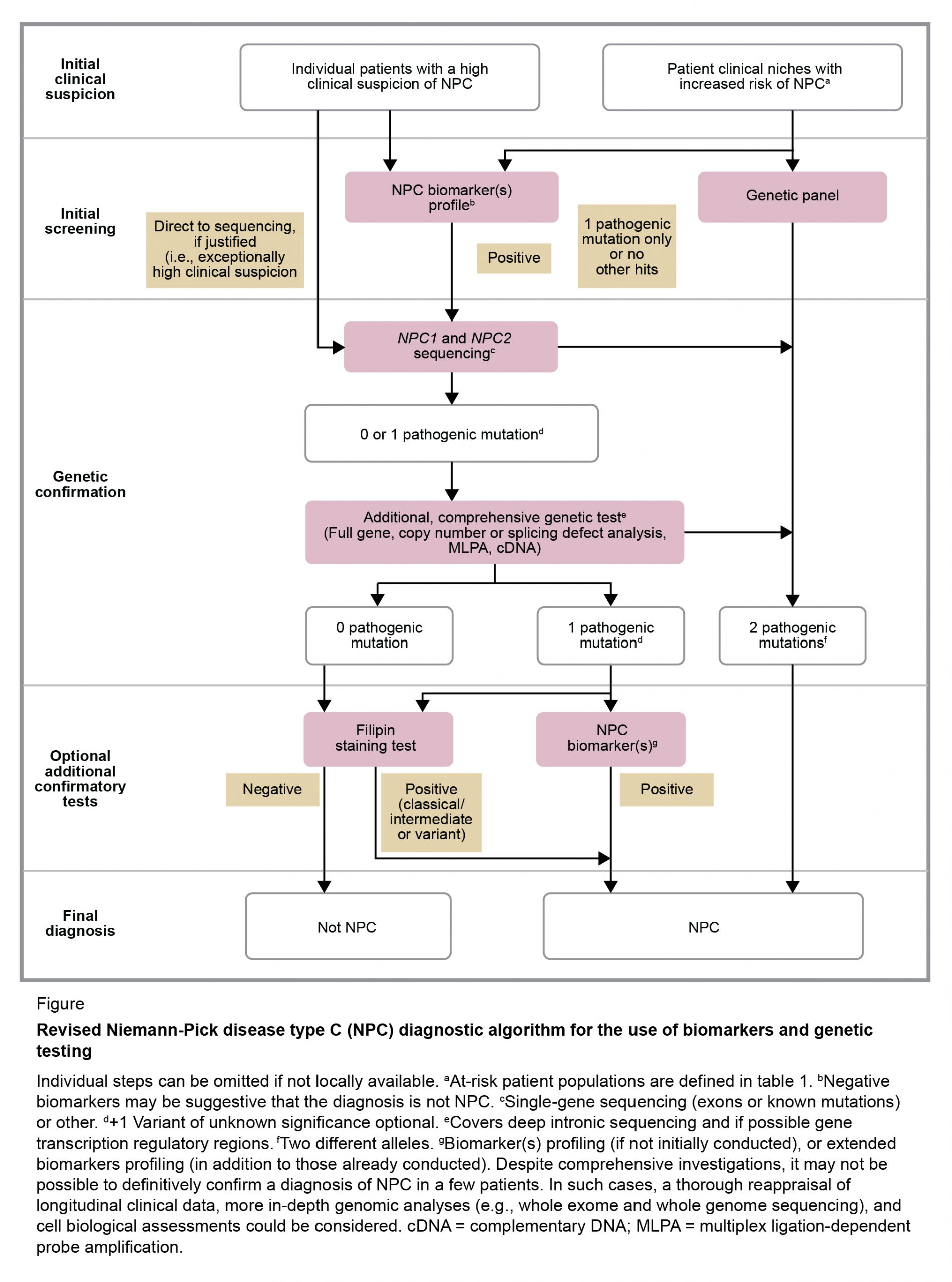
Current guidelines indicate that neuroimaging tests may be useful but are not diagnostic in patients with NPC. For instance, magnetic resonance imaging (MRI) of the brain may reveal atrophy in the frontal cortex and cerebellum, but this is not specific to NPC.[7]
MANAGEMENT AND CURRENT TREATMENT
Although there is no cure for NPC, many manifestations and symptoms of the disease are treatable. Because NPC affects many different organ systems and function, a multidisciplinary team should be involved in patient care (Table 2).[1] It is strongly recommended that patients with NPC receive evaluation and care at a medical center that has specific experience in this genetic disorder.
TABLE 2: MEMBERS OF THE MULTIDISCIPLINARY NPC CARE TEAM[1]
|
Primary Care Physician Neurologist Psychiatrist Neuropsychologist Speech Therapist Metabolic Diseases Specialist |
Pediatric Neuro-ophthalmologist Occupational and Physical Therapist Orthopedic Surgeon Gastroenterologist Nutritionist Genetic Counselor |
Symptomatic treatment can be quite effective, at least early in the progression of the disease. Seizures, cataplexy, spasticity, and hypersalivation, for example, can be managed with therapy.[1]
Miglustat, a drug that is approved to treat Gaucher disease, is a substrate reduction therapy that appears to modify the progression of NPC.[10] It is not approved in the US for the treatment of NPC, but it has been approved in the European Union and many other countries for the treatment of neurological manifestations of patients with NPC disease.[10]
The results of an international, multicenter registry study indicated that miglustat halted or attenuated disease progression in a significant portion of patients. Approximately 70% (n=153/217) of the continuous miglustat therapy population had improved or stable disease. Specifically, patients taking miglustat had stable or improved scores for the following domains: ambulation (68%; n=156/230), language (74%; n=170/230), manipulation (69.2%; 155/224), and swallowing (71.3%; 164/230).[10] Therapy was often discontinued (in 26% of the patient population receiving at least one dose and one follow-up visit) because of disease progression or death.[10]
PROGNOSIS
The prognosis of patients with NPC is dismal today, particularly for patients with early-onset disease. In a UK registry study,[11] all six children with neonatal onset died before reaching their third month.[11] Of the eight children with early-infantile onset, 100% died before their ninth birthday. In those with late-infantile onset, nearly 60% died at 13 years of age.
Nineteen of 42 with juvenile-onset NPC died at an average age of 26 years, compared with 8 of 25 in the oldest age-of-onset group (who died at a mean 34 years).[11] Although patients with late onset of NPC symptoms generally have longer life spans, they only rarely reach 40 years of age.[4,11]
CLINICAL TRIAL INFORMATION
A check of www.clinicaltrials.gov in July 2020 revealed 11 studies that were ongoing or actively recruiting patients with NPC. These studies included direct treatment of the disorder as well as symptomatic care.
FUTURE THERAPEUTIC DIRECTIONS
Several research avenues are being undertaken, including gene therapy.[16] One of the medical therapies that is furthest along is arimoclomol, a drug that was submitted for FDA approval earlier this year.[17] This agent increases the production of heat shock proteins, which is believed to help defective misfolded proteins and improve the function of lysosomes.[18]
Other earlier-stage medical therapies are aimed at curbing neurodegenerative progression and other symptoms. For example, the interim results of a phase 1 trial of intravenous hydroxypropyl-beta-cyclodextrin suggested that in treated patients, levels of tau, a neuron-specific biomarker of disease, was reduced in cerebrospinal fluid. Tau concentrations may be a biomarker for neurodegeneration associated with NPC. The National Institutes of Health are also sponsoring a phase 1/2 trial to determine whether combined intrathecal and intravenous administration of cyclodextrin can treat subacute liver disease in patients with NPC.[19] This agent, also known as VTS-270 (2-hydroxypropyl-β-cyclodextrin) is in phase 1/2 study for patients from 4 to 21 years.[20] This agent showed potential for slowing of progression on neurological, disease-specific outcomes measures in patients with NPC.[20]
An agent called IB1001 (N-acetyl-L-leucine) is currently being investigated to treat the symptoms of NPC and prove its neuroprotective effects in a multinational clinical trial.[21]
RESOURCES
National Niemann-Pick Disease Foundation
A non-profit, unbiased patient advocacy and family support organization dedicated to supporting and empowering patients and families affected by Niemann-Pick disease, through education, collaboration, and research.
National Organization for Rare Disorders (NORD)
Provides a unified voice for the 30 million people who wake up every day to fight the battle with a rare disease, including parents and caregivers.
REFERENCES
- Geberhiwot T, Moro A, Dardis A, et al. Consensus clinical management guidelines for Niemann–Pick disease type C. Orphanet J Rare Dis. 2018;13:50.
- Trendelenberg J, Vanier MT, Maza S, et al. Niemann–Pick type C disease in a 68-year-old patient. J Neurol Neurosurg Psychiatry. 2006;77:997-998.
- Pineda M, Mengel E, Jahnova H, et al. A suspicion index to aid screening of early-onset Niemann–Pick disease type C (NP-C). BMC Pediatrics. 2016:6:107.
- Bianconi SE, Hammond DI, Farhat NY, et al. Evaluation of age of death in Niemann-Pick disease, type C: Utility of disease support group websites to understand natural history. Mol Genet Metab. 2019;126:466-469.
- Rosenbaum A, Maxfield FR. Niemann–Pick type C disease: Molecular mechanisms and potential therapeutic approachs. J Neurochem. 2011;116:789-795.
- Patterson M. Niemann–Pick disease type C, in Adam MP, Ardinger HH, Pagon RA, et al (eds): Gene Reviews. Seattle, University of Washington, 2019.
- Patterson M, Clayton P, Gissen P, et al. Recommendations for the detection and diagnosis of Niemann-Pick disease type C. Neurol Clin Pract. 2017;7:499-511.
- Zavesca. European Medicines Agency 2012 https://www.ema.europa.eu/en/medicines/human/EPAR/zavesca. Accessed July 31, 2020.
- Pineda M, Walterfang M, Patterson MC. Miglustat in Niemann–Pick disease type C patients: A review. Orphanet J Rare Dis. 2018;13:140.
- Patterson M, Mengel E, Vanier MT, et al. Treatment outcomes following continuous miglustat therapy in patients with Niemann–Pick disease type C: A final report of the NPC registry. Orphanet J Rare Dis. 2020;15:104.
- Imrie J, Heptinstall L, Knight S, Strong K. Observational cohort study of the natural history of Niemann–Pick disease type C in the UK: A 5-year update from the UK clinical database. BMC Neurol. 2015;15:257. Doc: 10.1186/s12883-015-0511-1.
- Patterson MC. Niemann–Pick disease overview. National Neimann–Pick Disease Foundation Inc. September 9, 2016. https://nnpdf.org/overview/. Accessed July 2, 2020.
- Hammond N, Munkacsi AB, Sturley S. The complexity of a monogenic neurodegenerative disease: More than two decades of therapeutic driven research into Niemann-Pick type C disease. Biochim Biophys Acta Mol Cell Biol Lipids. 2019;1864:1109-1123. Doi: 10.1016/j.bbalip.2019.04.002
- National Organization of Rare Disorders: Niemann Pick Disease Type C. https://rarediseases.org/rare-diseases/niemann-pick-disease-type-c/ . Accessed November 5, 2020.
- Patterson M, Mengel E, Wijburg FA, et al. Disease and patient characteristics in NP-C patients: Findings from an international registry. Orphanet J Rare Dis. 2013;8:12.
- Amicus Therapeutics and the University of Pennsylvania announce major expansion of gene therapy collaboration (press release). Amicus Therapeutics May 29, 2019 https://ir.amicusrx.com/news-releases/news-release-details/amicus-therapeutics-and-university-pennsylvania-announce-major. Accessed August 1, 2020.
- Orphazyme completes rolling submission of new drug application to US FDA for arimoclomol for Niemann–Pick disease type C (press release). Orphazyme July 20, 2020. https://www.orphazyme.com/news-feed/2020/7/20/orphazyme-completes-rolling-submission-of-new-drug-application-to-us-fda-for-arimoclomol-for-niemann-pick-disease-type-c. Accessed August 1, 2020.
- Kirkegaard T, Gray J, Priestman DA, et al. Heat shock protein–based therapy as a potential candidate for treatment sphingolipioses. Sci Transl Med. 2016;8(355):355ra118. Doi:10.1126/scitranslmed.aad9823.
- Combined intrathecal and intravenous VTS-270 therapy for liver and neurological disease associated with Niemann-Pick disease, type C1. ClinicalTrials.gov January 31, 2020 https://clinicaltrials.gov/ct2/show/NCT03887533?term=NIH+study+19-CH-0028&draw=2&rank=1. Accessed July 29, 2020.
- Open-label Study of VTS-270 in participants with neurologic manifestations of Niemann-Pick Type C1. ClinicalTrials.gov July 15, 2020 https://clinicaltrials.gov/ct2/show/NCT03879655. Accessed July 29, 2020.
- IntraBio (press release) March 25, 2020. https://nnpdf.org/files/2020/03/IntraBio-FDA-Fast-Track-Designation-for-NPC-03242020.pdf. Accessed July 29, 2020.
This Niemann-Pick Disease Type C Learning Page is supported by Orphazyme. The content was developed by CheckRare and reviewed by Orphazyme.
Related Articles
How Families Cope with a New Rare Disease Diagnosis
Advice From a Niemann-Pick Disease Mom
Overview of Niemann-Pick Type C
Diagnosing Niemann-Pick Type C
Treatment Options for Niemann-Pick Type C
Advice to Parents of Children Diagnosed with Niemann-Pick Type C
How Advocacy Groups Support Patients with Niemann-Pick Type C
“The Fight is On”: Mother Argues for Continued Use of Experimental Drug for Niemann-Pick Disease